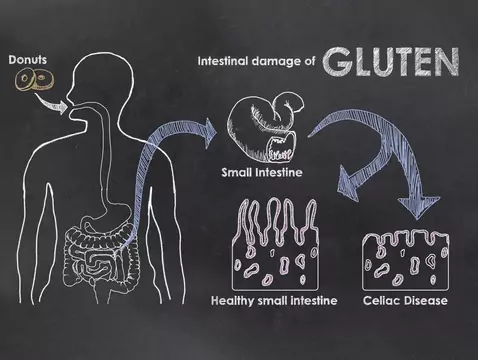
Rasmussen-Enzephalitis ist eine extrem seltene Krankheit. Leider ist die Krankheit aufgrund ihrer verstärkten Symptome und ihrer hohen Therapieresistenz äußerst schwer zu behandeln. Die einzige Möglichkeit, die Krankheit zu bestätigen, bleibt eine Hirnbiopsie, die zudem mit einem hohen Risiko für den Patienten verbunden ist. Bislang gibt es viele Mythen über die Ursachen für den Ausbruch der Krankheit, aber im Moment neigen Wissenschaftler dazu, Immunstörungen für den Ausbruch verantwortlich zu machen.
Werbung: